
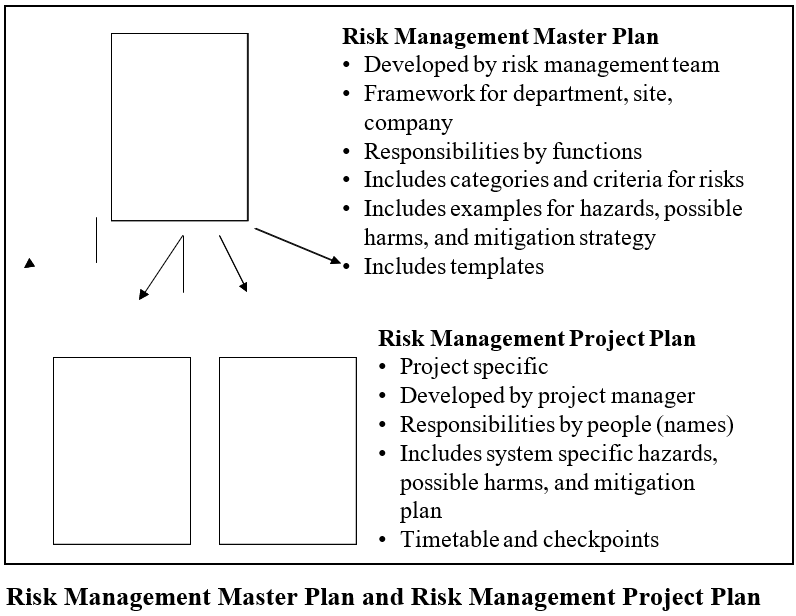
The Risk Management Master Plan aims to ensure efficient and uniform execution of risk assessment and control procedures throughout the organization, aligning with regulatory standards, customer expectations, quality benchmarks, and business objectives. It also emphasizes the importance of comprehension and adherence to the company’s risk management protocols across all levels of the organization.
This comprehensive plan serves as the foundation for individual Risk Management Project Plans, guiding specific risk management initiatives within the company.
The master plan contains descriptions about:
- The approach to the company’s risk management.
- Who in the organization is responsible for what.
- Other documents related to risk management plans.
- Which products and processes need risk management.
- Contents of individual Risk Management Project plan.
- How severity and probability are defined.
- Factors contributing to high and low severity.
- How to make a high-level risk assessment.
- Detailed steps for risk management.
Risk management starts when we first plan or buy a system, and it continues until we stop using it and move all important data to a new system.
We begin by quickly checking each system to see if it’s high, medium, or low risk. This helps us decide if we need a detailed risk management plan and which rules we should follow.
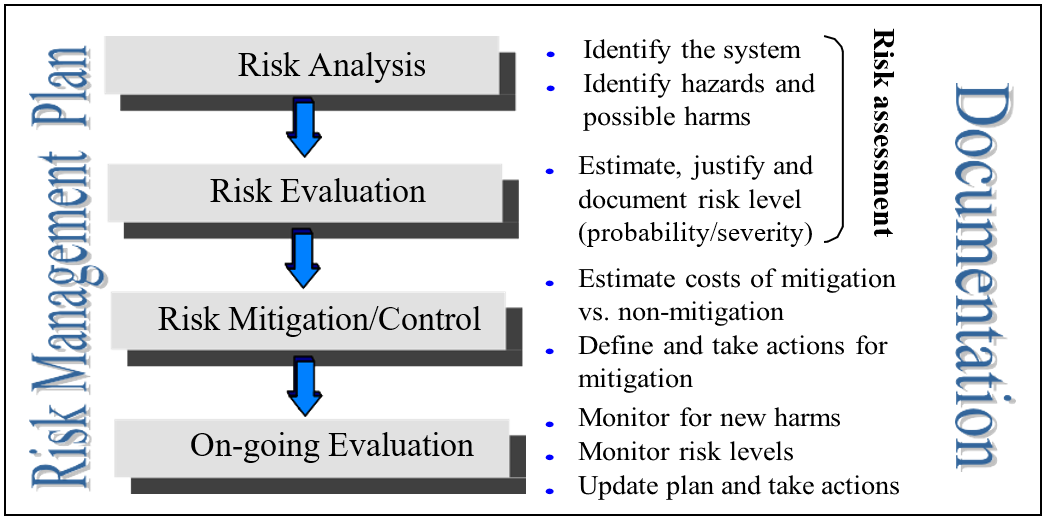
Systems that seem medium or high risk get a closer look. We break down this closer look into four steps, shown in below Figure :
Risk Management Master Plan and Risk Management Project Plan
While the Risk Management Master Plan is a framework and applicable to all projects, individual projects should be covered by Risk Management Project Plans (RMPP). This plan outlines risk management activities and should be specific to the project. The master plan should be used to draft this plan. The relationship between the master plan and the project plan is shown in below Figure.
Severity :
Severity in general means: how big is the problem if it occurs? Probability means: what is the likelihood that a problem occurs? Sometimes probability of detection has been suggested as an other measure. In the scope of this master plan probability of detection is included in ‘severity’. Severity factors get higher if the probability of detection is low.
Factors contributing to severity are : Impact on product Quality, Impact on People’s Health and Safety, Impact on Compliance and Business Continuity
Probability:
Probability shows the likelihood that the system can fail or generate wrong data or that data are lost? Probability should be expressed in occurrence per time. The categories can be :
- Frequent (once every month),
- probable (once in one to three months),
- improbable (once in three to twelve months),
- occasional (once in one to three years) and
- Impossible
It is important to use the levels for probability exactly as defined above or else redefine them. If they are redefined the definition should be documented. We use past experiences from the same or similar systems to estimate the probability. We use the same form as shown in table 1 and then add probability. From overall severity and probability we can then calculate the overall risk expressed by risk codes 3 (High), 2 (Medium) or 1 (Low).
High-Level Risk Management
Many times we need to make a rough risk estimation for a process or system, for example, when we need to decide if and to what extent Part 11 requirements should be implemented. The assessment can be done by following these five steps:
- Describe the system, where it is used and what it
- Define a couple of key factors as to why a system could fail, what could cause the failure and what the consequences could
- Define severity of consequences in case the system
- Give a description of when and how many times a system has failed. Define probability that a system may
- Make a final risk assessment and classify the system and data generated by the system as high, medium and or low
Detailed Risk Management :
Detailed risk management should be done:
- If there are no or insufficient data present to make a reasonable risk assessment based on high-level inputs as explained in the previous chapter.
- If the high-level risk assessment results in a high or medium risk system and we need to develop a risk mitigation plan to reduce the risk to an acceptable level. In this case a detailed risk analysis with factors contributing to risks should be identified together with hazards and possible harms and their level of severity and probability.
Steps for detailed risk management include risk identification and analysis, risk evaluation, risk mitigation and on-going evaluation and control.
A. Risk Analysis :
The first step in the risk management process is the risk analysis, sometimes also called risk identification or Preliminary Hazard Analysis (PHA). The output of this phase is the input for risk evaluation. Inputs for risk analysis are:
- Intuitive science or engineering sense
- Specifications of equipment/hardware/software
- Users experience with the same system already installed
- Users experience with similar systems
- IT staff experience with the same or similar systems
- Experience with the vendor of the system
- Failure rates of the same or similar systems
- Trends of failures
- Validation reports
- Service records and trends
- Internal and external audit results
- FDA inspection reports and warning letters
Inputs can come from operators, the validation group, IT administrators or from QA personal, e.g., as a result of an audit or findings from internal and external FDA inspections.
B. Risk Evaluation:
After information on risks has been collected, the risk evaluation phase starts. In this phase the risk management team takes information from the risk identification form, goes through each individual risk that has been described and defines levels for severity and probability. Numbers are associated to the levels and the overall risk factor R is calculated using the formula:
R = (H x 2 + B) x O
H = Health/Safety risk, B = Business continuity risk, O = Occurrence, R = Risk factor
Multiplication of H by a factor of two means that health/safety has a higher impact than risks associated with business impact. Resulting risk factors can range from 3 to 45. The risk factors are further categorized into risk codes 1 (High if R>20), 2 (Medium if R=11-20) and 3 (Low if R<10).
C. Risk Mitigation:
This phase evaluates whether or not the risks as estimated in the previous phase are acceptable. If they are not acceptable, alternatives on how to mitigate risks should be evaluated and implemented.
The risks should be put into three categories :
Factor 10 and lower: Code 1 – Routinely accepted, no action taken.
Factor 11-20: Code 2 – Operation requires written, time limited waiver, endorsed by management. Mitigation subject to cost/benefit analysis.
Factor higher than 20: Code 3 – Not accepted. Mitigation required. Alternative approaches should be evaluated.
It is important to estimate the cost for mitigation as well as potential losses through non-mitigation. Losses for non-mitigation should include real or direct costs and tangible or indirect costs. Real costs are loss of revenue and are relatively easy to estimate. Tangible costs are more difficult to estimate. They can include damage of a company reputation or getting onto the FDA’s radar screen after one or more failed FDA inspections.
There are different ways and approaches to mitigate risks. They can range from process and system design changes to the training of existing staff. Typically the most effective ones are also the most expensive and take the longest time.
Design and Process Changes : This could be a new type of equipment, a new functionality or a process change. A typical example of a process change would be to complement the quality control analysis of a final product with in-process analysis and control. Typically this results in new equipment with consequent costs for purchasing, installation, validation and familiarization. It also requires development of new SOP’s and training.
Extended Testing/Validation: This does not remove the product problem but shows weaknesses of the system and conditions under which the system fails can be identified. This only helps if procedures and work around solutions can be developed and implemented to avoid known critical conditions. A typical example would be to test a system under increasing high load and to always operate the system below the point when it started to fail in the test phase.
Extended Monitoring and Warnings : This is useful when the results are graphically presented and warning and action limits have been defined. Operators should have clear instructions on what to do if critical limits are approached.
Better People Qualification: The idea here is to avoid risk situations that are caused by human errors. Alternatives are to better train existing staff or to hire new people who have more experience with the same or similar systems. Compared to other approaches it can be quite cost effective and mitigation can be achieved in a relatively short time if existing people can be trained. However, it can become expensive and take longer if new people have to be hired.
The initial and on-going costs for the best alternative should be estimated and compared with the estimated costs of non-mitigation. For risk codes B (= medium risk) this comparison should be the basis for the decision whether to mitigate or not. The rational behind the decision should be well justified and documented.
Once the decision to mitigate has been made and the strategy is identified, a plan should be developed with the owner regarding the time schedule and deliverables.
D. Ongoing Monitoring, Review and Control:
Once the plan is in place and the system is running, the effectiveness of the plan should be monitored, reviewed and adjusted if necessary. The monitoring program should also help to identify previously unrecognized hazards. These could have been introduced by changing processes or introducing new technologies.
The monitoring program should check if risk factors have changed to either higher or lower values. Users and IT professionals gain a lot of experience that can be used to further optimize the effectiveness. If factors exceed the previous specified limit, mitigation strategies should be evaluated. If higher values decrease below the threshold, mitigation may not be necessary anymore which can save operating costs.
Contributors document observations and make recommendations. They should also make a recommendation if the change should be implemented urgently if it is time critical. The risk management team evaluates the recommendations made by contributors. The team meets monthly if there are no recommendations for changes classified as ‘urgent’. In the case of an urgent recommendation for a change the team meets and evaluates the change within a week.
E. Documentation:
Regulatory agencies strongly suggest basing decisions for details of compliance with GxP’s and 21 CFR Part 11 on a justified and documented risk assessment. Therefore documentation is very important.
Documentation is also important to justify investments as needed to meet business requirements. Complete documentation for risk management should include:
- Risk Management Master Plan : This shows your company’s approach towards risk assessment and risk management.
- Risk Project Management Plan: This shows the plan for specific system and mitigation strategies.
- Lists with description of risk categories, ranking criteria and results of ranking.
- Justification for non-mitigating risks with high factors.
- Risk mitigation plans
- Mitigation actions taken
- Review reports
Related Articles :
How to do Risk Management of Computer Systems Used for FDA Compliance
How to Do Risk Assessment of IT Networks for FDA compliance
How to do risk assessment of Spreadsheet and Macros
Risk management of existing(Legacy) systems for FDA Compliance